What is a metadata table for RNA-Seq data? And why is it important?
What is metadata?
Metadata is a type of data that describes corresponding data that is independent of the data itself. However, metadata should not be considered a subcategory of “data”. “Metadata” to one researcher, could be another’s “data”. Whether data is considered “data” or “metadata” depends on how it is being used. When it comes to RNA-Seq experiments, the metadata will describe the samples the RNA was isolated from. It might be helpful to identify all experimental variables in your experiment when documenting metadata. As our computational biologist advises:
“Metadata includes information about source of the samples, like the organism, cell line, tissue, developmental stage; conditions of the experiment like treatment or dose; health status such as whether samples were healthy or diseased. If they were diseased, what stage the disease was; It can also describe about the wet lab experiment and library preparation methods”
Types of metadata

Types of metadata according to FAIR and Sabot et al. (1,2)
When thinking of the metadata to document for your sequencing experiment, it may be helpful to understand different types of metadata to ensure your documentation is thorough.
According to FAIR, metadata can be categorized into 3 types: (1)
Administrative metadata are data concerning the management and administration of resources related to your project. Think of things like your funding source, your PI, and any project collaborators you have.
Descriptive or citation metadata are data that identify the dataset itself. For example, think of what information you would use to find a dataset on the NIH’s National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO). You would likely use descriptive metadata such as authors, title, keywords, or related publications.
Structural metadata is data concerning how the dataset was obtained and structured. Out of the three types, structural metadata is the most important for understanding a dataset and how to reproduce it. This category describes how your samples was collected, the experimental variables & values, number of samples, etc. Think of what you would detail in your methods section in your paper for your sequencing experiment; these would all fall under structural metadata.
Sabot et al offers an alternative way to categorize metadata that is presented as more specific to genomic research. (2) Although organized differently, these still cover the same information as FAIR metadata types.
Sampling metadata refers to where your DNA or RNA sequences came from. This could refer to a location, a human subject, an animal model, a cell line, etc. Think back to your last sequencing experiment, where did your DNA/RNA samples originate from?
Handling metadata is any information surrounding how the data was obtained, that is distinct from how the data was analyzed. Therefore, this is concerned with methodology such as what protocols or instruments you used to collect data. Sabot’s handling metadata is synonymous with FAIR’s structural metadata.
Processing metadata refers to any analyses or transformation the dataset has undergone. This is important, as it could affect any subsequent interpretations or analyses performed.
Why is metadata important?
Metadata is important for enabling scientific advancement and discovery associated with data sharing. Reliable and accurate metadata supports transparency, validation, exploration, and reproducibility of results. (3) Metadata is also crucial for the reuse of NGS data, in either meta-analyses or multi-omics studies. (4) Open access, for data and publications, is trending globally and here to stay. To be truly open access and reap the benefits of data sharing, providing metadata is indispensable.
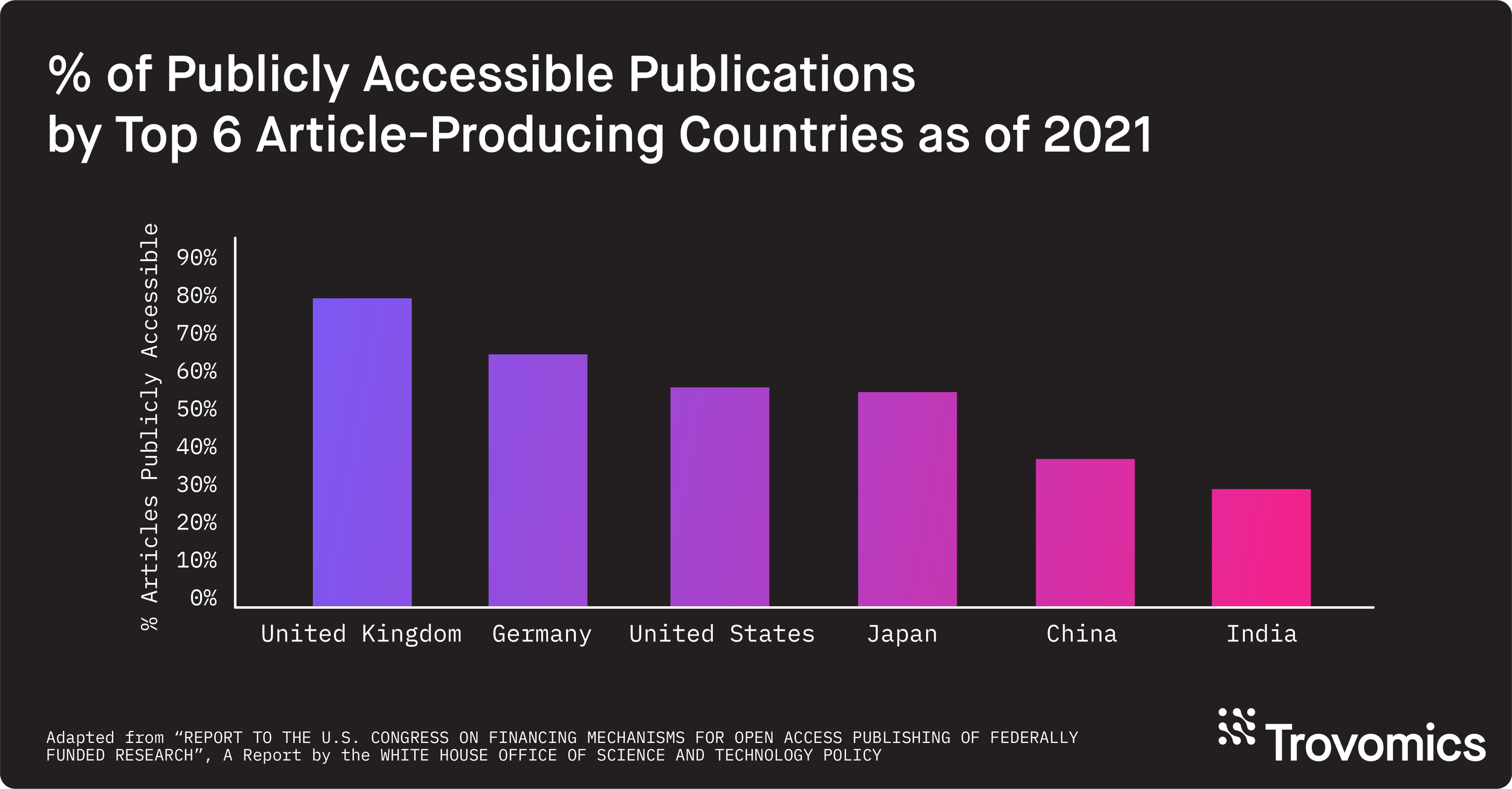
As of 2021, over 50% of all publications in the US, Germany, Japan, and United Kingdom are publicly available. (5) In parallel, the number of open access journals increases yearly. (5) Major publishers of academic research recognize the benefits associated with data sharing and transparency. Publishers will often strongly endorse or require sharing of the data associated with an original research article at the time of publication. Additionally, if your research is NIH-funded, you are subject to their Data Sharing and Management (DSM) Policy, as well as their Genomic Data Sharing (GDS) Policy if your research produces genomic data. In summary, not only is metadata important for scientific rigor and reproducibility, but also for complying with academic publishers’ and funding institutions’ policies.
For more information on the FAIR Guiding Principles, read Wilkinson et al. or check out How to Fair. (1,6)
NIH-funded research containing genomic data is required to be made available through any widely used data repository. With NCBI GEO as the largest publicly available repository for high-throughput gene expression data (array- and sequence-based), it is likely you may choose GEO as your repository. When submitting data to GEO, it is encouraged to adhere to the Minimum Information about a high-throughput SEQuencing Experiment (MINSEQE) guidelines set forth by the Functional Genomics Data Society. (7) However, these are good guidelines to follow when documenting metadata in your own use as well. MINSEQE describes essential five elements of experimental information to provide, specifically when releasing high-throughput sequencing (HTS) data: (7)
“The description of the biological system, samples, and the experimental variables being studied: "compound" and "dose" in dose-response experiments or "antibody" in ChIP-Seq experiments, the organism, tissue, and the treatment(s) applied.”
“The sequence read data for each assay: read sequences and base-level quality scores for each assay; FASTQ format is recommended, with a description of the scale used for quality scores.”
Sequencing services typically provide sequencing read data as FASTQ files, which includes both read sequences and quality scores. FASTQ is the standard format for storage and widely accepted for downstream analyses. Indeed, at Trovomics, we accept your sequence read data as FASTQ files.
“The ‘final’ processed (or summary) data for the set of assays in the study: the data on which the conclusions in the related publication are based, and descriptions of the data format.”
“General information about the experiment and sample-data relationships: a summary of the experiment and its goals, contact information, any associated publication, and a table specifying sample-data relationships.”
“Essential experimental and data processing protocols: how the nucleic acid samples were isolated, purified and processed prior to sequencing, a summary of the instrumentation used, library preparation strategy, labelling and amplification methodologies, alignment algorithms and data filtering plus data processing & analysis protocols.”
In summary, there are efforts such as FAIR and MINSEQE that aim to uphold high-quality metadata standards for sequencing experiments. Regardless of which guidelines you follow, we recommend thinking not only about your experimental design (variables, hypothesis, etc), but also beyond that:
Where did your samples come from? Are they from cells, tissue, animals, or human subjects?
How would you identify your samples? What are their genotypes or phenotypes?
What experimental variables were you testing? What was different between your samples?
Were there any notable events that occurred at any point in your experiment?
What did you write in your lab notebook during your experiment?
What protocols did you use?
Did you perform any transformations to your data after it was collected?
Any of these answers would qualify as metadata. From here, you will be able to extract the metadata you need to meet the minimum requirements of a repository, journal, or institution.
How is a metadata used for RNA-Seq analysis?
Metadata in RNA-Seq analysis are used to determine what comparisons to make in downstream analyses, such as identifying differentially expressed genes. It will also influence how your results are visualized. Going by FAIR metadata types, RNA-Seq analysis is primarily concerned with structural metadata. Without metadata, there is no way for an analysis software to know if a FASTQ file was sequences from a sample that was in your control or experimental group. As computational tools are used for downstream analysis and visualization, it is important to structure and format your metadata in a manner that is organized and systematic.

Simplified illustration of how metadata impacts RNA-Seq analysis and visualization.
Why are metadata organized in a metadata table for RNA-Seq?
For both storage and downstream analyses of RNA-Seq data, metadata are organized in a table called a metadata table. Metadata tables are typically saved in .CSV file format, so that it can easily be read and extracted by computer programs and scripts. Trovomics accepts your metadata in a .CSV format.
“The metadata table describes the experiment’s data in a detailed and unambiguous way, so that we should be able to clearly see the experimental design from it.”
How do you organize metadata in a metadata table?
The column titles in your metadata table will provide details about what metadata each column contains, such as sample name, file name, treatment, sequencing platform etc. Each row will represent one sample and designate its attributes. We recommended you keep a “master” metadata table of your RNA-Seq experiments for documentation and storage. As different downstream analyses may not need all types of metadata, a separate metadata table can be derived from your master.
“I include all data I have from the experiment, including sample IDs, sample source, experimental conditions, wet lab and library preparation methods. Also, adding a column for the groups to compare helps to define which comparisons we’d like run differential expression analysis for.”
Here is an example of a metadata table for a RNA-Seq submission to NCBI GEO:

NCBI GEO’s metadata table template
In this example, we see there is metadata documented for library name, title, organism, cell line, cell type, genotype, etc of the samples. Each row represents one sample and will contain specific values pertaining to that sample for each column. Notice, the same conventions are used throughout the entire metadata table (i.e., same capitalizations, spellings, abbreviations). This is also important for machines and other instruments to be able to accurately categorize all samples. For example, you wouldn’t want samples grouped separately as “WT” or “wildtype”, when they should all be categorized in the same group.
For Trovomics’s automated RNA-Seq pipeline to run successfully, we require the following metadata, at minimum, to be in a metadata table:
Sample Name
File Name
Experimental Variable(s) (≥1)
Here is an example of a metadata table for a RNA-Seq experiment in Trovomics:

Trovomics’s metadata table template
In conclusion, “metadata” in RNA-Seq describes the samples that the RNA was isolated from. Metadata are required for data transparency, validation, reproducibility, and reuse. Metadata for RNA-Seq analysis are organized in a table format so that both the researcher and the computer can clearly discern the experimental design from it. Trovomics, and other RNA-Seq analysis pipelines, will take this experimental design to specify which samples belong to each of the groups to compare in differential expression, and to create the plots to visualize the results.
Interested in making a metadata table?
References:
Metadata - How to FAIR. https://howtofair.dk/how-to-fair/metadata/.
Sabot, F. On the importance of metadata when sharing and opening data. BMC Genom Data 23, 79 (2022).
Puntambekar, S., Hesselberth, J. R., Riemondy, K. A. & Fu, R. Cell-level metadata are indispensable for documenting single-cell sequencing datasets. PLoS Biol. 19, e3001077 (2021).
Stevens, I. et al. Ten simple rules for annotating sequencing experiments. PLoS Comput. Biol. 16, e1008260 (2020).
OSTP. REPORT TO THE U.S. CONGRESSON FINANCING MECHANISMSFOR OPEN ACCESS PUBLISHINGOF FEDERALLY FUNDEDRESEARCH. https://www.whitehouse.gov/wp-content/uploads/2023/11/Open-Access-Publishing-of-Scientific-Research.pdf (2023).
Wilkinson, M. D. et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016)
FGED Society. Minimum Information about a high-throughput SEQuencing Experiment. MINSEQE https://www.fged.org/projects/minseqe.